![]()
![]()
![]() Previous: Predictions of flux tube model
Up: Introduction
Next: Equations of motion
Previous: Predictions of flux tube model
Up: Introduction
Next: Equations of motion
A molecular dynamics simulation is a method with a simple approach: take the initial condition equations one wishes to solve, and apply numerical integration techniques to them. Usually, the problem is not tractable by analytical techniques because of the large number of particles involved.
Traditionally, molecular dynamics simulations are applied to very large numbers of particles, often with ill-defined, or very complicated interaction equations. A typical application is fluid flow simulations:
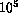 )
)
Point three is not true in the flux tube simulation: the direction of the force depends on the pairings. The optimal pairing is not a local function, but is globally determined. This global nature of the forces makes the problem computationally more difficult: a large molecular dynamics simulation can often otherwise be broken up into many smaller pieces, with each piece computed in parallel.
One property of the flux-tube simulation is that the desired
simulation size is not large. A simulation of a large atomic nucleus
(say U-238), would involve 238 nucleons, or 714 quarks. As will be
seen later,
the classical mesonic simulation should go as 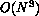 (
(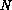 being the number of quarks).
The baryonic simulation, however, requires
being the number of quarks).
The baryonic simulation, however, requires 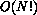 time.
time.
[1] and [10] provide good arguments for designing heuristics that do not require complete enumeration of all combinations. These heuristics were not applied because of the limitation to mesonic matter.
mcr@ccs.carleton.ca